-

ALL RESULTS
-

CADD
-

PROTEIN PRODUCTION AND TARGET BASED ASSAYS
-

PHENOTYPIC SCREEN
-
STRUCTURAL BIOLOGY
-

GENOMICS
-

MECHANISM OF ACTION
-

AUTOMATED HOMOLOGY MODELING WORKFLOW FOR DRUG TARGET
-
PRODUCTION AND TUNING ON HPC INFRASTRUCTURE
-

REGULATORY CONTRACTS
-
DISSEMINATION & EXPLOITATION OF THE FOREGROUND
CADD
Summary
Generation of homology models based on the mapped functional proteins of the entire sequenced SARS-CoV-2 viral genome
The sequences of mature proteins were determined from the genome and annotations from UniProt.
The SWISS-MODEL platform was used to generate homology models. Possible heteromeric complexes were predicted and modeled as well. The resulting models, as well as experimentally determined structures deposited in the PDB for the SARS-CoV-2 proteins, are available on a dedicated page of the SWISS-MODEL server. The page is updated on a weekly basis with the latest structures from the PDB and improved models.
STRUCTURAL BIOLOGY
Summary
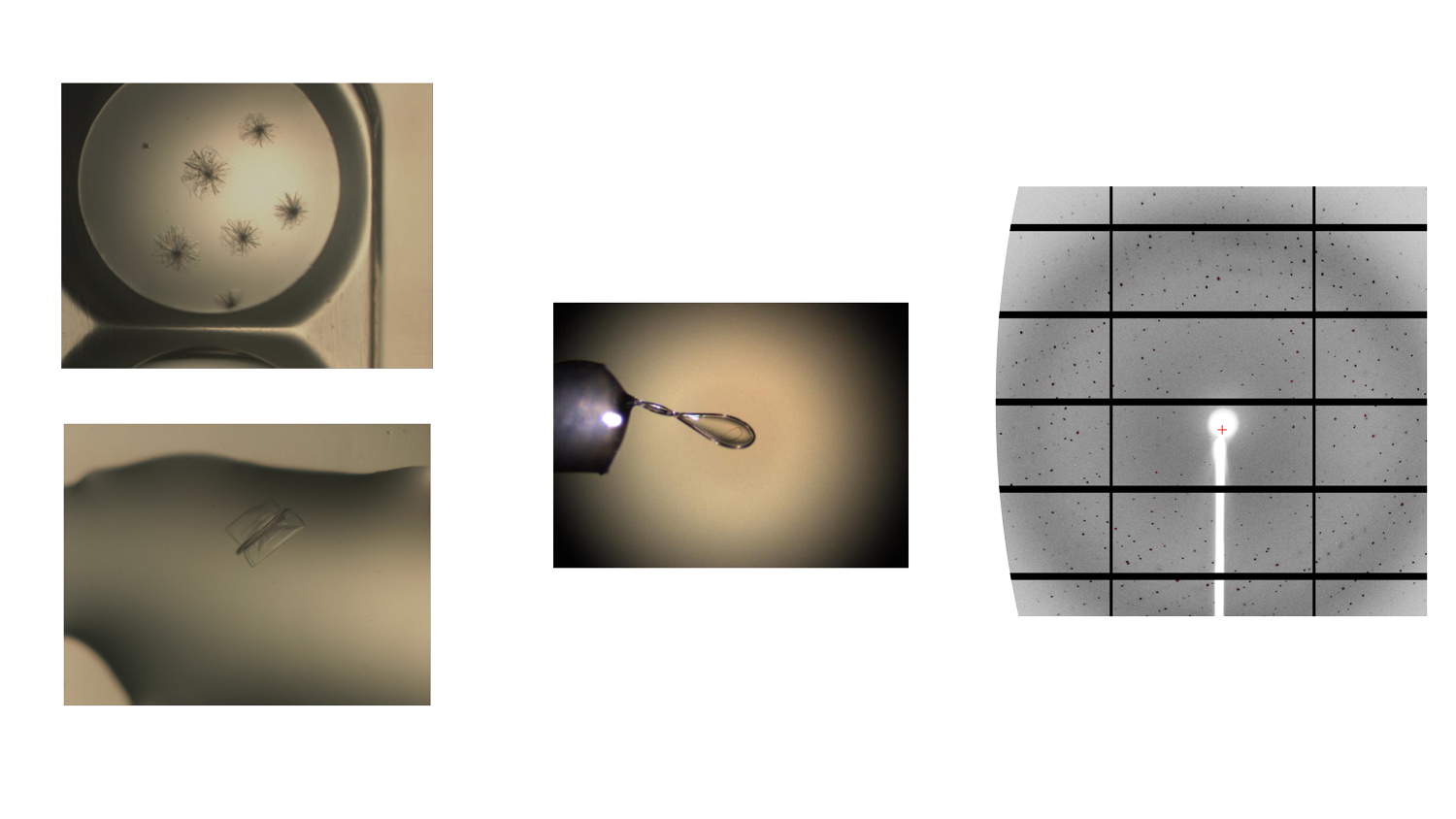
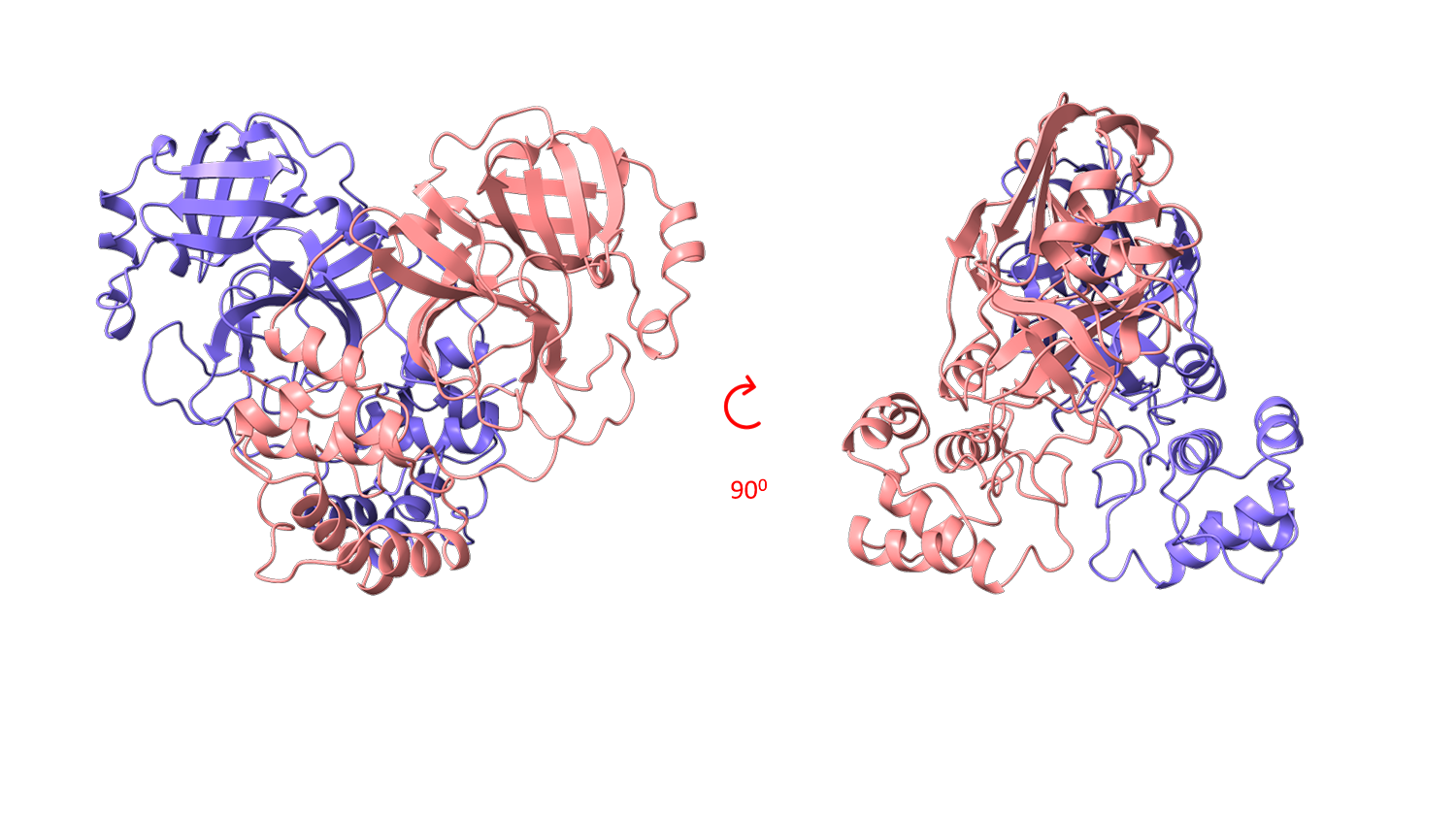
The structural biology team of Elettra Sincrotrone Trieste has focused on SARS-CoV-2 Mpro viral protein by setting up a reproducible expression, purification and crystallization protocol and using thermal stability profiling to define protein quality control and batch to batch reproducibility. Thousands of protein crystals of the APO and HOLO forms have been obtained, and tested at the XRD2 beam line of Elettra. Best crystals diffracted at a resolution of 1.3 Å. Hundreds of data set have been collected, processed and refined and resulting 3D models confirmed the Mpro dimeric structure of with its typical fold.
Figure 1 shows examples of Mpro crystals and diffraction patterns, Figures 2 and 3 show the 3D graphic representations of the Mpro dimer.
A few more details
The structural biology team of Elettra Sincrotrone Trieste involved in the WP4, started to work in April 2020, anticipating the planned activities by about 5 months with respect to the original schedules. The initial effort has been dedicated to protein expression, purification and sample characterization with crystallization screening set-up.
The SARS-CoV-2 Mpro was successfully expressed in E.coli and purified at homogeneity (> 98% purity) starting from the expression vector kindly donated by L. Hilgenfeld (Lubeck University – DE). Initial crystallization screenings have been set up based on published data and using commercially available crystallization kits. The most promising conditions have been optimized and “flower-like” shape crystals were reproducibly obtained. These crystals were cryo-preserved and tested at the XRD2 beamline of the Elettra synchrotron, showing a diffraction pattern with a resolution in the range of 1.6-2.0 Å, with the best resolution achieved at 1.52Å. Datasets were processed and a quick MR solution was done using pdb 6W63 as a starting model showing either P21 or C2 space group, having a monomer or a dimer in the asymmetric unit, respectively, but with almost identical conformation. Further optimization of the crystallization trials has established also a seeding based protocol that grew crystals of space group P212121. For each space group we obtained the crystal structure of Mpro in APO form and the coordinates have been deposited in PDB with code 7ALI (https://www.rcsb.org/structure/7ALI, sp.gr. P21), 7ALH (www.rcsb.org/structure/7ALH, sp.gr. C2) and 7BBE (www.rcsb.org/structure/7BB2, sp.gr. P2(1)2(1)2(1)).
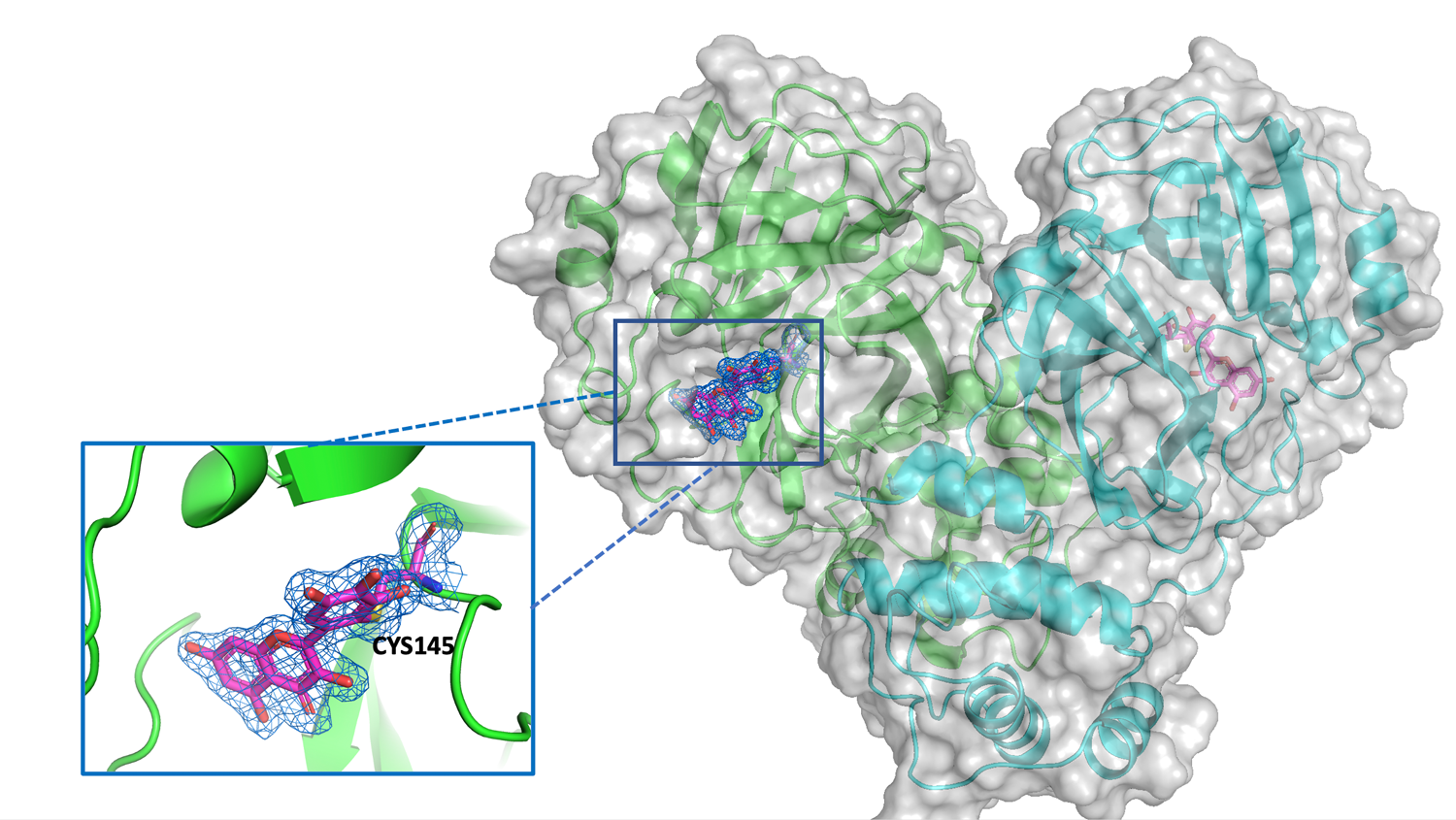
Xray diffraction studies on crystals of Mpro bound to specific inhibitors have been performed. A number of small molecules selected from a biochemical screening of 8700 repurposing drugs have been tested by co-crystallization and soaking techniques. So far 8 structures have been deposited in PDB describing the binding modes of myricetin, Ebselen, SU3327, MG-132 in absence and in presence of DTT. Binding mode of myricetin, a flavonoid, has been clearly identified in the crystal structure (Fig. 4) showing a covalent linkage with the catalytic cys145. The results have been published in two papers confirming the findings of the biochemical screening [Kuzikov et al. 2021; doi.org/10.1021/acsptsci.0c00216] and validating the outputs of virtual screening and molecular dinamics [Gossen et al. 2021; https://doi.org/10.1021/acsptsci.0c00215]. The coordinates of the crystal model are deposited in PDB with code 7B3E (www.rcsb.org/structure/7B3E). All structures in PDB have been released immediately and are openly available.
Deposited PDB:7ALH, 7AL, 7B3E, 7BB2, 7BE7, 7BFB, 7BGP, 7NBY, 7NF5, 7NG3, 7NG6
PRODUCTION AND TUNING ON HPC INFRASTRUCTURE
Summary
In the WP8 “Production and tuning on HPC infrastructure”, the POLIMI team is responsible for the continuous tuning and code adaptation of the EXSCALATE platform, and contributes to its porting on the new Marconi100 partition at CINECA.
The first month of the E4C project coincided with the initial production period of the new CINECA machine. POLIMI ported a first version of the docking library to exploit the computing capability of the heterogeneous computing node composed of 2xIBM-Power9 Sockets and 4xNVidiaV100.
During the experimental campaign, we reached a throughput of more than 250K optimal ligand poses per second on a single node of the Marconi100 machine. Porting and tuning on multiple nodes is currently ongoing.
A few more details
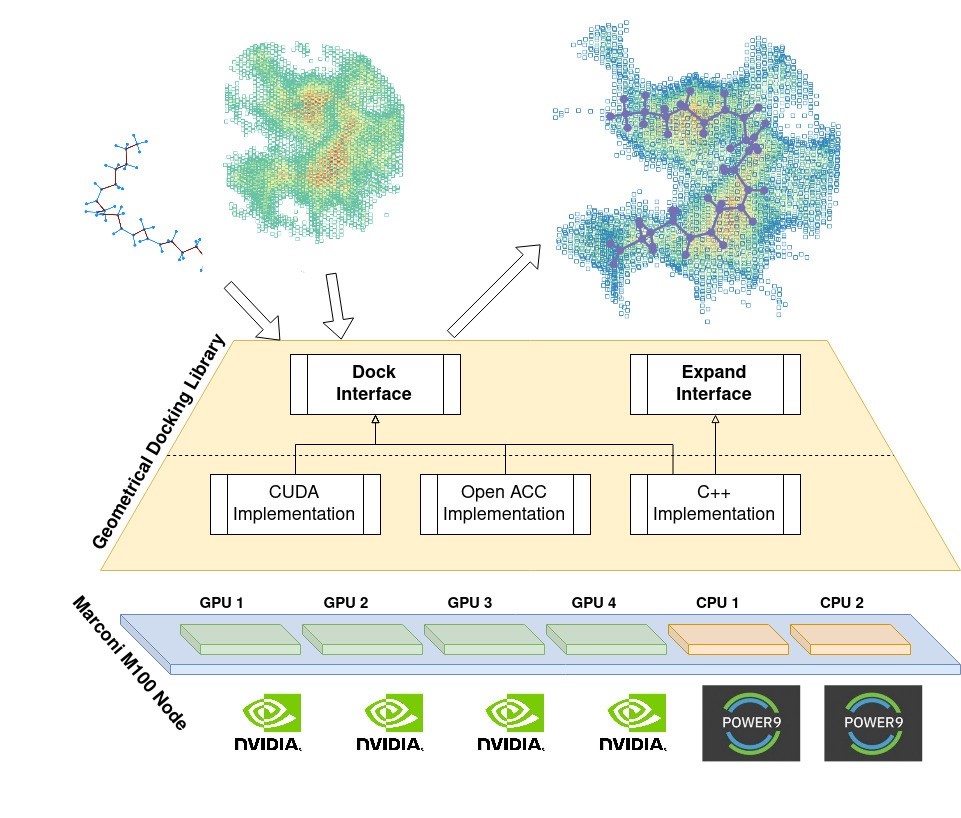
To promote agile and portable software development needed to guarantee continuous releases of updated functionalities and improved elaborated throughput of the EXSCALATE software platform, we encapsulated all the geometrical docking, issued in a stand-alone library called LiGen GeoDock, with a simple, stable, and well-defined non-virtual interface.
Figure 1 shows an overview of the LiGen GeoDock library, which aims at docking a ligand in a target pocket using geometrical information only. To better encapsulate Geodock in the EXSCALATE platform, we designed a single public interface hiding implementation details. In this way, we can improve GeoDock without hindering the development of the other components. The E4C project targets a heterogeneous node composed of GPUs and CPUs. The previous C++ code version was unable to harness the full computational power of the new CINECA Marconi100 node based on CPUS only. To overcome this limitation, we implemented the docking algorithm using the OpenACC pragma-based language and the PGI OpenACC compiler to generate device-specific code while maximizing the code portability. However, given the transition to the Marconi100 supercomputing cluster at CINECA, we then implemented the docking algorithm using the CUDA C/C++ language to obtain the maximum performance from of the NVIDIA Tesla V100 GPUs.
We ran an initial experimental campaign to assess the ligand library performance with a wide range in the number of atoms and rotatable bonds. The code peak performance using only the 2xIBM-Power9 and 128 software threads reaches a throughput of 30K optimal ligand poses per second, while the exploitation of the full node including the 4xNvidiaV100 GPUs reaches a throughput of more than 250K optimal ligand poses per second.
These performance results obtained so far on the new CINECA Marconi100 partition are very promising, and they are a good starting point for further tuning of the LiGen GeoDock library. As next steps, we envision pushing further on a fine-grain refinement and adopting dynamic autotuning approaches, combined with the entire machine-level scaling.
CADD
Summary
MD simulations of the HM generated, and on the 3D experimental structure deposited in the Protein Data Bank (D1.2).
We proceeded to simulate MD simulations of the homology models generated, and on the 3D experimental structure deposited in the Protein Data Bank. The production run was performed to generate at least 1 μs (1 microsecond) trajectory with a total of 20,000 collected structures for each simulated system. The viral protein dataset, selected for MD simulation studies, contains Active Interest Proteins and Low-Interest Proteins in their in apo form. To select the most useful protein conformation form MD, a post HPC-run analysis was performed by using different clustering methods.
A few more details
Reported below is the dataset containing viral proteins, selected for MD simulation studies, and that contains Active Interest Proteins and Low-Interest Proteins:
| M-Protein | N-Protein | |
| Nsp2 | Nsp3 | |
| Nsp4 | Nsp5 - 3CL-PRO | |
| Nsp6 | Nsp7-Nsp8 - HETEROMER | |
| Nsp9 | Nsp12 - MONOMER | |
| Nsp12-Nsp7-Nsp8 - HETEROMER | Nsp13 - HELICASE | |
| Nsp14 - MONOMER | Nsp10-Nsp14 - HETEROMER | |
| Nsp15 | Nsp16 - MONOMER | |
| Nsp10-Nsp16 - HETEROMER | ORF3a | |
| ORF6 | ORF7a | |
| ORF8 | ORF10 | |
| PL-PRO | Spike-ACE2 | |
| Spike |
Among these 25 unique structures, our studies have used both Homology and Experimental models, by increasing the number of overall structures to 37. All the MD simulations, carried out on HPC5 and Galileo clusters, yielded by ENI and CINECA respectively, are ongoing, and 27 systems already reached at least 1 microsecond, and among these, some have reached or exceeded 2 microseconds. We are pushing the simulation times towards 10 micro-seconds. In particular, the 3CL-PRO was simulated in its DIMERIC and MONOMERIC forms, to better understand which are the most important structural differences. The analysis carried out, with the work-flow that will be explained in the next paragraphs, allowed acquisition of useful information that will be collected in a scientific work.
Due to the high interest of the scientific community on this target, we have already produced a manuscript entitled “Computational Studies of SARS-Covid2 3CLpro: Insights from MD Simulations”, that will be submitted to the International Journal of Molecular Sciences - Special Issue “Exscalate4CoV: Innovative High Performing Computing (HPC) Strategies to Tackle Pandemic Crisis” in a few days. In this paper we discuss the main differences coming from the analysis of the whole protein structural behavior and those seen in the binding site. The web address of the repository will be communicated shortly. It will be possible to download the trajectories of the simulations discussed in the work.
Papain-like proteinase (PL-PRO): Responsible for the cleavages located at the N-terminus of the replicase polyprotein. In addition, PL-PRO possesses a deubiquitinating/deISGylating activity and processes both 'Lys-48'- and 'Lys-63'-linked polyubiquitin chains from cellular substrates. Participates, together with nsp4, in the assembly of virally induced cytoplasmic double-membrane vesicles necessary for viral replication. In the video, the PL-PRO is shown in green highlight.
Nsp12-7-8: A key component, RNA-dependent RNA polymerase [RdRp, also known as nsp12], catalyzes the synthesis of viral RNA, and therefore plays a central role in the replication and transcription cycle of the COVID-19 virus, possibly with the help of nsp7 and nsp8 as cofactors. In the video, the nsp12/nsp7/nsp8 hetero-oligomeric complex is shown in highlight. Nsp12, nsp7 and nsp8 are shown in blue, red and green respectively.
<Nsp13: Scientists suspect that nsp13 unwinds so that other proteins can read its sequence and make new copies. This protein, called Helicase, is a multi-functional protein with a zinc-binding domain in the N-terminus displaying RNA and DNA duplex-unwinding activities with 5' to 3' polarity. Activity of helicase is dependent on magnesium. Here, the protein is reported in blue highlight.
<Nsp15: This enzyme is a specific endoribonuclease with a C-terminal catalytic domain, belonging to the EndoU family. EndoU enzymes are present in all animal kingdoms, where they perform various biological functions associated with RNA processing. Researchers suspect that this protein cuts the residual virus RNA as a way of hiding from the antiviral defenses of the infected cell. The protein, in its hexameric form, is shown in the video shows in highlight style, and each monomer composing the hexamer has a different color.
Spike receptor-binding domain (RBD)/ACE2: Dynamic structure of the receptor-binding domain (RBD) of the spike protein of SARS-CoV-2 bound to the cell receptor ACE2. Coronaviruses use the spike glycoprotein on the envelope to bind to their cellular receptors. Such binding triggers a cascade of events that leads to the fusion between cell and viral membranes for cell entry. The video shows in highlight, the SARS-CoV-2 RBD core in slate and ACE2 in red.
CADD
Summary
A systematic mapping of the druggable cavities within the SARS CoV-2 therapeutically relevant proteins.
This study provided a novel strategy for pocket-mapping based on the combination of pocket (as performed by the well-known FPocket tool) and docking searches (as performed by PLANTS or AutoDock/Vina engines). Such a mapping enables the identification of the most relevant binding sites for which virtual screening simulations or de novo rational design should allow the identification of promising hits.
A few more details
Such an approach is implemented by the Pockets2.0 plugin for the VEGA suite of programs. The VEGA suite comprises a graphical interface with a new version of the plug-in for FPocket (named Pockets2.0), a well-known software used to detect protein cavities, based on an optimized algorithm for Voronoi tessellation. For a better exploration of the protein cavities, this combines the already implemented cavity mapping, as performed by Fpocket, with docking calculations with probe molecule(s) using AutoDock/Vina or PLANTS docking programs. To optimize the ranking of the explored cavities, Pockets2.0 can utilize both Fpocket and docking scores by calculating customizable consensus scores. The combination of the FPocket and docking scores by calculating customizable consensus scores leads to a significant increase of the correctly identified binding sites compared to the FPocket and docking scores alone, and this enhancement appears to be truly relevant when analyzing complex proteins with rather narrow binding pockets, and in particular, for characterizing allosteric binding sites.
The scientific paper entitled “A systematic mapping of the druggable cavities within the SARS CoV-2 therapeutically relevant proteins by combining pocket and docking searches as implemented in Pockets2.0”has been submitted to International Journal of Molecular Sciences - Special Issue “Exscalate4CoV: Innovative High Performing Computing (HPC) Strategies to Tackle Pandemic Crisis”. The web address of the repository will be communicated shortly. It will be possible to download all the structural data discussed in the work.
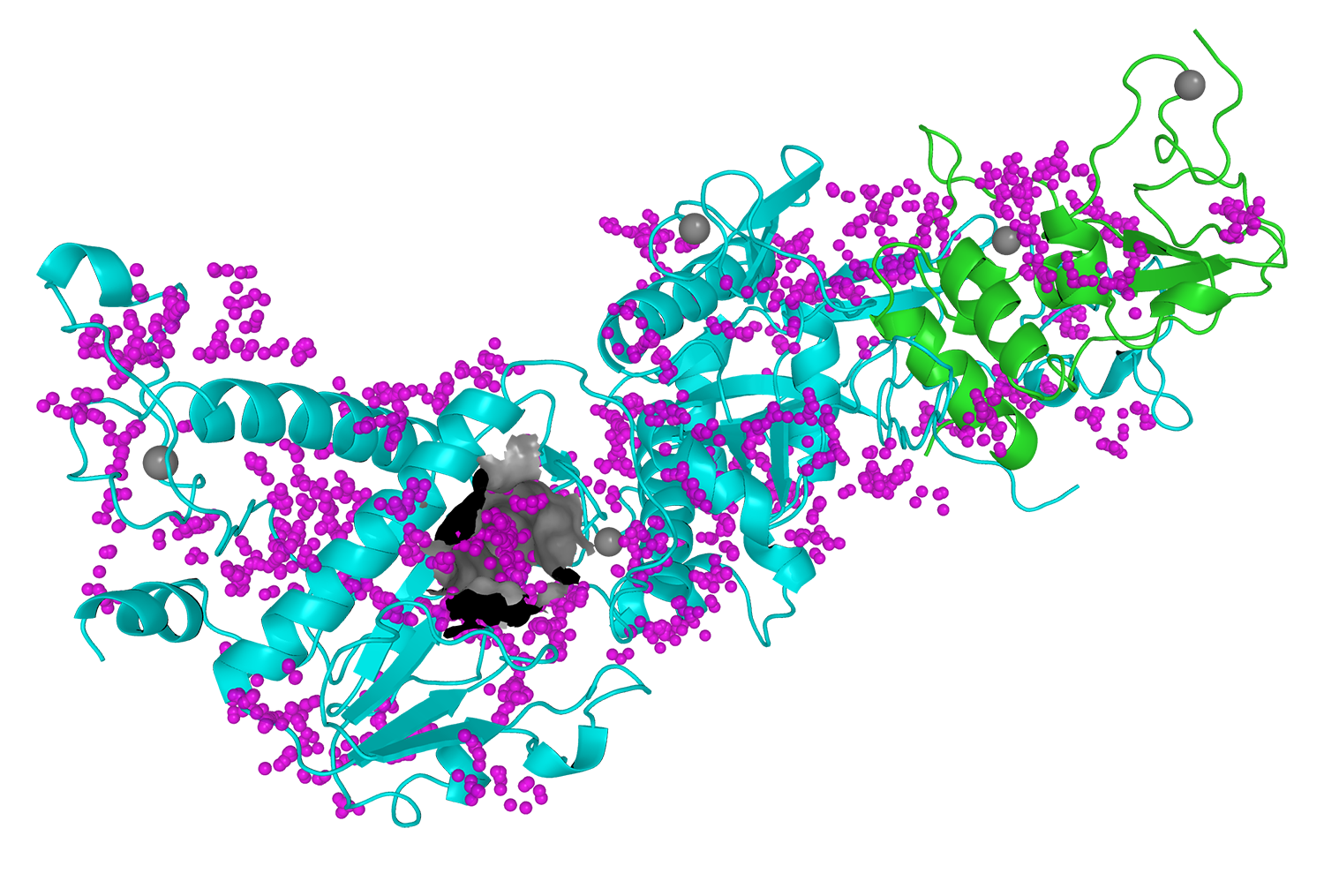
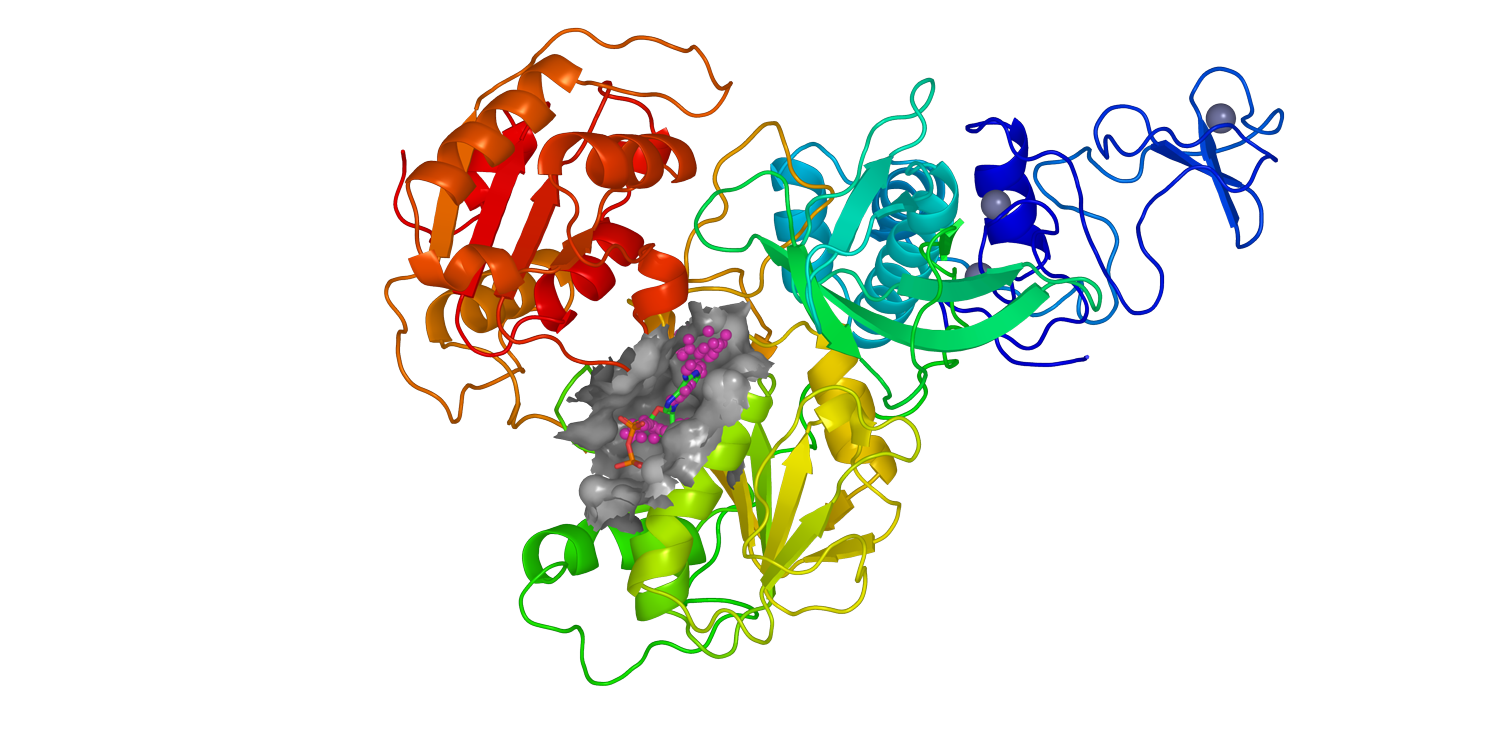
The images below show the process of the viral protein mapping (Figure 1) and the identification of the binding pocket (Figure 2). In particular, figures 1 and 2 represent the homologic 3D structures of the viral protein nsp13 and the heteromer nsp14-nsp10 respectively, generated in the deliverable D1.1. This process underlines the importance of collaboration among the activities carried out by each partner.
CADD
Summary
Machine Learning and Virtual Screening (VS) protocol optimization and Drug Repositioning (D1.3, D1.4, D1.5).
Several docking simulations were performed to define and optimize the machine learning and virtual screening protocols to use on SARS CoV-2 proteins. The performances of the virtual screening strategies were assessed by evaluating their capacity to correctly rank molecules, which are endowed with antiviral activity, and in particular, with a known effect against SARS CoV proteins, considering the lack of known actives on SARS CoV-2 proteins.
The tuned and validated virtual screening protocols were used to screen a repurposing library, containing the set of safe in man drugs, commercialized or under active development in clinical phases, and a set of known bioactives in particular preclinical compounds identified as “CoV Inhibitors” (> 12000 drugs). The most promising drugs, identified from docking studies as potentially active against SARS-Cov-2 proteins, were selected to be tested in biochemical and phenotypic assays performed respectively in WP2 and WP3.
A few more details
The SARS CoV-2 3CL-Protease was analyzed and used to fine tune the virtual screening strategies. SARS CoV 3CL-Protease known inhibitors were collected from literature to define training sets for our studies, keeping in mind that the pocket of SARS-Cov and SARS-Cov-2 proteases are very similar. Different docking algorithms were combined to increase the quality of the virtual screening campaign.
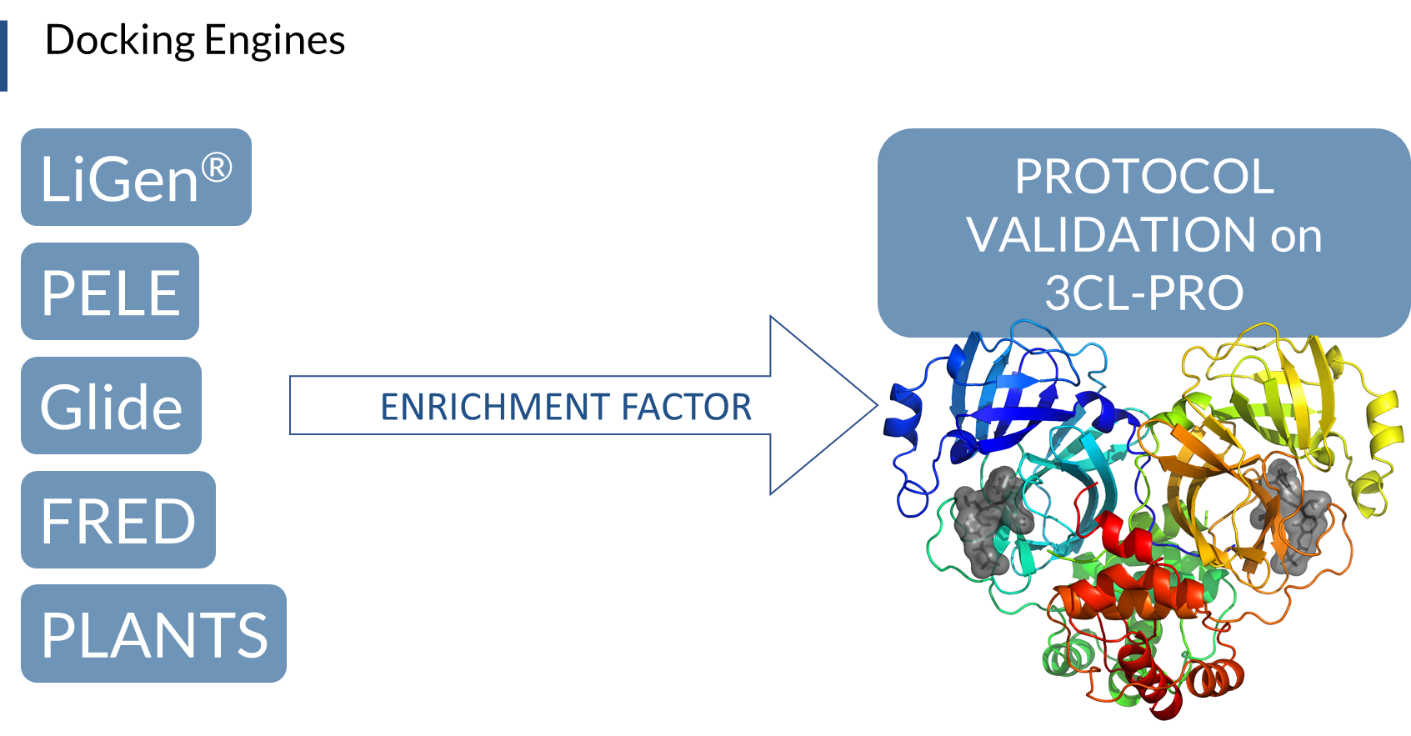
Five consortium partners (Dompé, University of Milan, and the three supercomputing centers, CINECA, Barcelona Supercomputing Center (BSC) and Jeulich Forschungszentrum (FZJ)), made their hardware and software facilities available to run docking simulations, including LiGen, PLANTS, PELE, Glide and FRED. The virtual screening protocols were validated in terms of Enrichment Factor, setting the conditions that allow most of the training set compounds to be ranked in the top-scored positions. Subsequently, the molecules with the best docking scores, found in the top 1% of the screened library, were selected with each software, and a consensus approach of the different docking scores was used to rank the most promising safe in man drugs acting on 3CL-PRO viral protein (Figure 1). To further improve the quality of the results, more accurate methods were applied, such as induced-fit simulations on SARS-CoV known inhibitors with PELE software, and a regression analysis model to correlate predicted and in vitro activities was built. We obtained a clear correlation of higher IC50 with larger residence time in the active site, which was analyzed in terms of (1) the solvent-accessible surface area of the ligands, (2) the sub-pocket population analysis. Both analyses showed high potential for a more accurate prediction, opening the possibilities, for example, to a second refinement effort after an initial docking campaign.
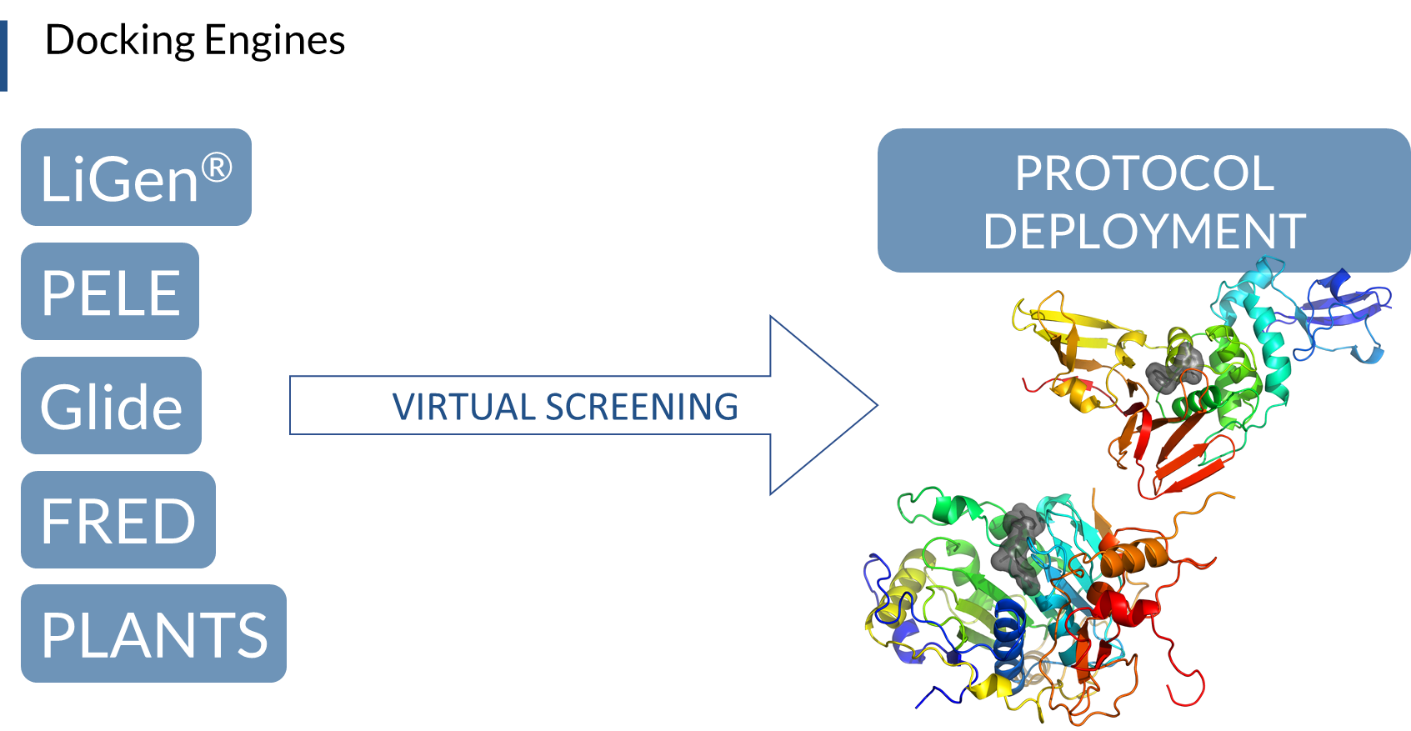
We are now using the tuned and validated virtual screening protocols to screen the drug repurposing library against the best selected 3D structures of the other SARS-Cov-2 proteins, either already experimentally obtained (PDB) or derived by homology modelling techniques, and processed by molecular dynamics simulation (Figure 2).
We currently have validation compounds coming from the literature, and results coming from our docking approaches that identified different sets of molecules. We obtained a critical assessment of different approaches and results that we can get from combining the best of the techniques we have available and that we are putting in place, for the identification of the most promising safe in man drugs that are ready for immediate treatment of the infected population.
DISSEMINATION & EXPLOITATION OF THE FOREGROUND
Summary
Setup and Maintenance of Website & Social Media (D10.1).
Chelonia leads the coordination of dissemination and communication activities. The project webpage www.exscalate4cov.eu was produced within the first month of the project and is now regularly updated and maintained. Communication activities ensure the diffusion of the project outcome outside the Consortium so that the methodologies developed and the results obtained can be widely disseminated in the scientific community.
Communication objectives are achieved by using multiple different strategies including the use of social networks such as Linkedin, Facebook and Twitter to spread the project knowledge to a larger audience.
Dedicated exscalate4cov pages have been created and all communication are mainly tagged #exscalate4cov. Given the wide social impact of this research, actions towards the general press are given through press releases, newsletters and through local press.
DISSEMINATION & EXPLOITATION OF THE FOREGROUND
Summary
Initial Dissemination Activities and Plans (D10.2).
This task defines the strategy for exploitation of the project results. The activity, led by CHELONIA, in tight collaboration with DOMPE, analyzes the strategies and implements the actions to maximize the utilization of the project results, beyond the project partners and time-frame. A dissemination plan was issued to all partners 3 months earlier than the expected deadline in order to facilitate dissemination and to align partners in communicating projects results to the target audience. A first webinar was organized on April 28th, 2020, thanks to the support of CECAM (Centre Européen de Calcul Atomique et Moléculaire – www.cecam.org ), which is an organization devoted to the promotion of fundamental research on advanced computational methods and to their application to important problems in frontier areas of science and technology. To access the recorded webinar please visit YOUTUBE
REGULATORY CONTRACTS
Raloxifene
Raloxifene has emerged from the Exscalate platform as a molecule with potential effectiveness against SARS-CoV-2, based on antiviral targeting. This molecule is used as active ingredient in medicines already authorized on the European Community for the treatment of post-menopausal osteoporosis. A preliminary development and registration strategy was shared with the European Medicines Agency (EMA) in October 2020 through a procedure called Rapid Scientific Adivice.
The product is currently being used in a placebo-controlled Phase 2/3 clinical study, which is ongoing in Europe.
PROTEIN PRODUCTION AND TARGET BASED ASSAYS
Summary
Identification of inhibitors of SARS-CoV-2 3CL-Pro enzymatic activity using a repurposing screen (“target confirmation of main protease”)
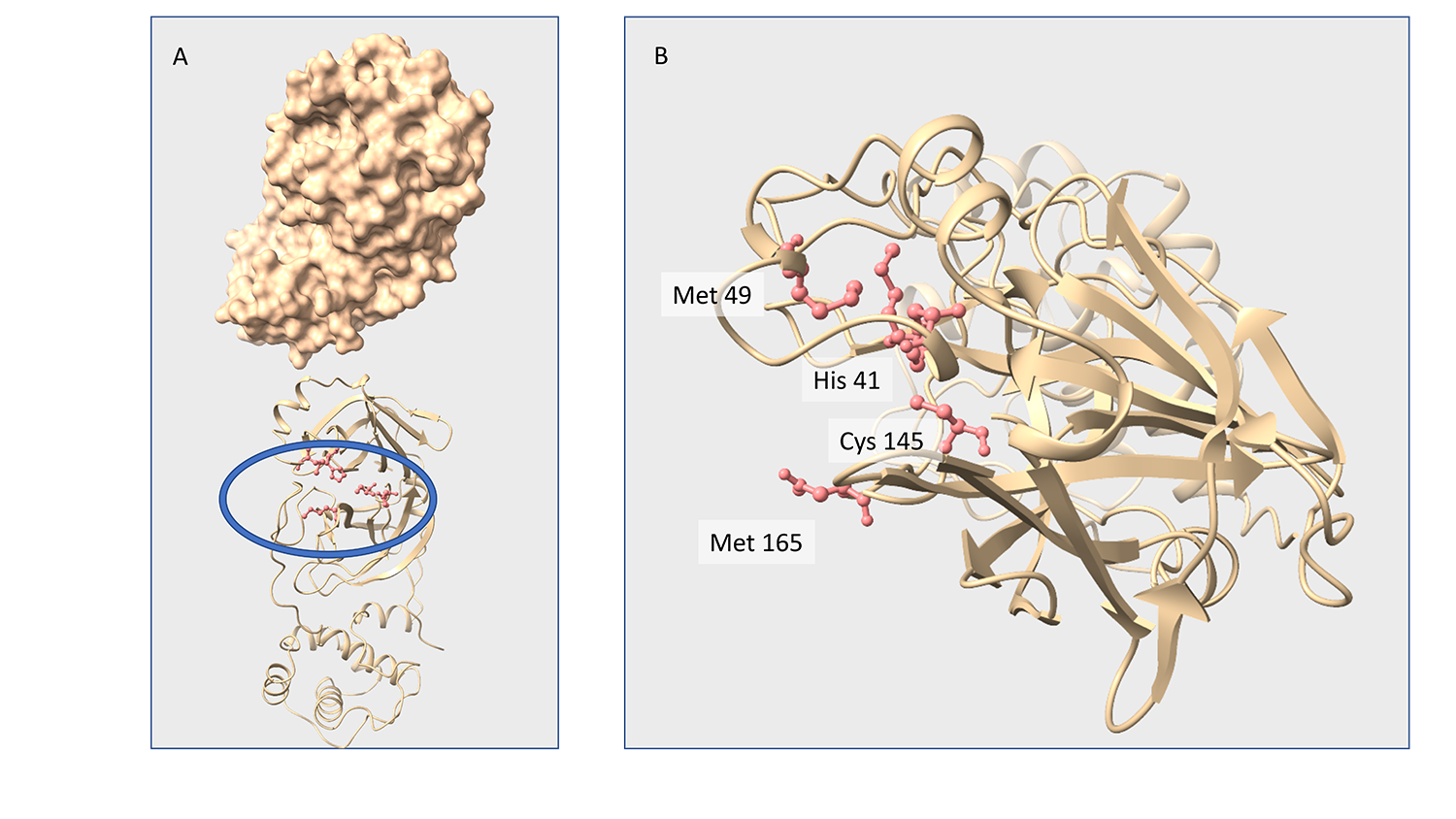
SARS-CoV-2 main protease (3CL-Pro), also termed M-Pro, is an attractive drug target as it plays a central role in viral replication by processing the viral polyproteins pp1a and pp1ab at multiple distinct cleavage sites. Within our repurposing program we confirmed previously reported inhibitors of 3CL-Pro and have identified 62 additional compounds with IC50 values below 1 μM. A subset of eight inhibitors showed anticytopathic effect in a Vero-E6 cell line. , and the X-ray crystal structure of the complex of one of the inhibitors and SARS-Cov-2 3CL-Pro was solved, showing covalent binding to the catalytic Cys145.
PROTEIN PRODUCTION AND TARGET BASED ASSAYS
Summary
X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease (“How do you find 37 new repurposed drugs that have direct 3D binding to SARSCoV2's main protease to knock out its ability to replicate”)
In a search for a drug against COVID-19, we have performed a high-throughput X-ray crystallographic screen of two repurposing drug libraries against the SARS-CoV-2 main protease (Mpro), which is essential for viral replication. In contrast to commonly applied X-ray fragment screening experiments with molecules of low complexity, our screen tested already approved drugs and drugs in clinical trials. From the three-dimensional protein structures, we identified 37 compounds that bind to Mpro. In subsequent cell-based viral reduction assays, one peptidomimetic and six non-peptidic compounds showed antiviral activity at non-toxic concentrations. We identified two allosteric binding sites representing attractive targets for drug development against SARS-CoV-2.103 authors mainly from science organizations in Hamburg contributed to this work; 5 of our ITMP scientists contributed scientific input and data interpretation and managed to supply our compounds to this X-ray crystallography screening.
GENOMICS
Summary
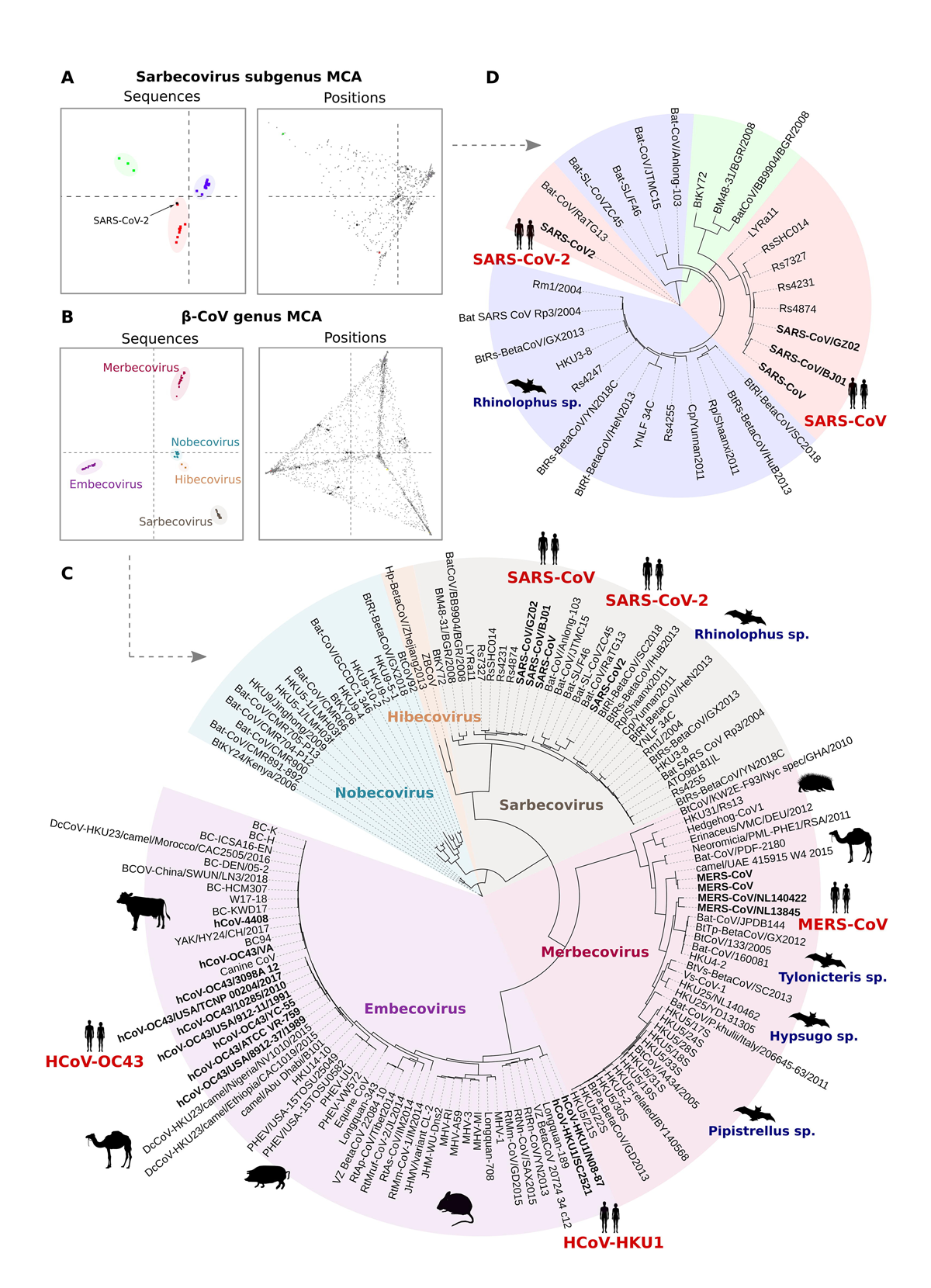
Main activities performed in WP5 include the use of techniques for molecular evolution, phylogenetic and multiple correspondence analysis (MCA) for the identification of genomic and protein sites associated with SARS-CoV-2 adaptation and host-pathogen interaction.
As a first step, the analysis focused on the spike protein from SARS-CoV-2 and other representative β-Coronaviruses. Our results shows that the unsupervised analysis of conservation patterns across the β-CoV spike protein family can help tracing the amino acid space encoding the specificity of β-CoVs to their cognate host cell receptors. More precisely, our results indicate that host cell receptor usage is encoded in the amino acid sequences of different CoV spike proteins in the form of a set of specificity determining positions (SDPs)1.
A few more details
Here, we aim at analyzing the effect of evolutionary constraints in shaping the organization of the spike protein family across the β-CoV lineage. To this aim, we use a multivariate analysis protocol for unsupervised detection of both the protein family segregation and the residue positions that better explain the sources of variation of the family, i.e. Specificity Determining Positions (SDPs).
The analysis was performed both at the level of the full β-CoV genus and individual subgroups. While in the first case the results are consistent with the known phylogenetic classification of Betacoronavirus into five subgenera (Fig. 1C), the analysis performed on the individual β-CoV groups allowed a fine-grained separation into clusters that clearly reflect the functional diversification of the spike protein family, that is, the specificity to different host-cell receptors. Indeed, both the clustering and domain enrichment results of the identified SDPs consistently reproduce the known cell receptor specificities observed across the different β-CoV lineages. At the level of the Sarbecovirus group, for example, both SARS-CoV-2 and RaTG13 are clustered together with SARS-CoV and other SARS-like sequences from bats (Fig. 1A,D), reflecting the ability of these members of the Sarbecovirus group to bind the ACE2 cell receptor. Notably, the proximity of SARS-CoV-2 and RaTG13 and other SARS-CoV sequences based on the SDPs is different to what observed based on phylogenetic analysis, where they form distinct clades.
Collectively, our results point to a key role of the SDPs in mediating host cell receptor specificity across β-CoVs and provide, at the same time, a framework for monitoring the evolution of the SARS-CoV-2 specificity to hACE2, as well as the emergence of novel potential cross-species transmission events. As such, it is worth mentioning that from the analysis of amino acid variations across the circulating SARS-CoV-2 virus, SDPs found in the RBD tend to mutate with a very low frequency, similar to what is seen at ACE2 contacting sites. This is of relevance, as our results suggest that mutations in SDPs can significantly impact the receptor-binding ability of the spike. In line with this reasoning, it is important to notice that other members of the Sarbecovirus group might have the potential to acquire ACE2 binding ability, as they share substantial similarity in terms of SPDs. This is especially the case of members of the Sarbecovirus clade 3, which despite being phylogenetically distant to SARS-CoV-2, display identical residues in 10 out of 14 SDP positions within the RBD, making them potential candidates for new human infections.
In summary, our results show that the identification of evolutionary patterns based on the analysis of sequence information alone can provide meaningful insights on the molecular basis of host-pathogen interactions and adaptation. Both the methodology and results obtained in this work lead the basis for a follow-up study where, in collaboration with other members of the E4C consortium, we aim at identifying potential routes of mutations that could lead to new β-CoVs adaptation to human hosts, and ultimately contribute to better understanding and monitoring of events that are critical to public health concerns worldwide.
1Pontes C, Ruiz-Serra V, Lepore R, Valencia A. Unraveling the molecular basis of host cell receptor usage in SARS-CoV-2 and other human pathogenic β-CoVs. Comput Struct Biotechnol J. 2021;19:759-766. doi: 10.1016/j.csbj.2021.01.006
In parallel, continuous monitoring of the virus evolution is performed based on the analysis of the SARS-CoV-2 genomic sequences from public repositories. Variation analysis is performed based on the established SARS-CoV-2 NC_045512.17 as a reference genome, where amino acid variations are annotated for all major coding regions as well as alternative reading frames and mutation frequencies computed globally and by different countries at different time points. Data are continuously updated as new sequences become available and shared with the partners and the community via the ViralSeq website
CADD
Summary
Generation of homology models based on the mapped functional proteins of the entire sequenced SARS-CoV-2 viral genome
The sequences of mature proteins were determined from the genome and annotations from UniProt.
The SWISS-MODEL platform was used to generate homology models. Possible heteromeric complexes were predicted and modeled as well. The resulting models, as well as experimentally determined structures deposited in the PDB for the SARS-CoV-2 proteins, are available on a dedicated page of the SWISS-MODEL server. The page is updated on a weekly basis with the latest structures from the PDB and improved models.
CADD
Summary
MD simulations of the HM generated, and on the 3D experimental structure deposited in the Protein Data Bank (D1.2).
We proceeded to simulate MD simulations of the homology models generated, and on the 3D experimental structure deposited in the Protein Data Bank. The production run was performed to generate at least 1 μs (1 microsecond) trajectory with a total of 20,000 collected structures for each simulated system. The viral protein dataset, selected for MD simulation studies, contains Active Interest Proteins and Low-Interest Proteins in their in apo form. To select the most useful protein conformation form MD, a post HPC-run analysis was performed by using different clustering methods.
A few more details
Reported below is the dataset containing viral proteins, selected for MD simulation studies, and that contains Active Interest Proteins and Low-Interest Proteins:
| M-Protein | N-Protein | |
| Nsp2 | Nsp3 | |
| Nsp4 | Nsp5 - 3CL-PRO | |
| Nsp6 | Nsp7-Nsp8 - HETEROMER | |
| Nsp9 | Nsp12 - MONOMER | |
| Nsp12-Nsp7-Nsp8 - HETEROMER | Nsp13 - HELICASE | |
| Nsp14 - MONOMER | Nsp10-Nsp14 - HETEROMER | |
| Nsp15 | Nsp16 - MONOMER | |
| Nsp10-Nsp16 - HETEROMER | ORF3a | |
| ORF6 | ORF7a | |
| ORF8 | ORF10 | |
| PL-PRO | Spike-ACE2 | |
| Spike |
Among these 25 unique structures, our studies have used both Homology and Experimental models, by increasing the number of overall structures to 37. All the MD simulations, carried out on HPC5 and Galileo clusters, yielded by ENI and CINECA respectively, are ongoing, and 27 systems already reached at least 1 microsecond, and among these, some have reached or exceeded 2 microseconds. We are pushing the simulation times towards 10 micro-seconds. In particular, the 3CL-PRO was simulated in its DIMERIC and MONOMERIC forms, to better understand which are the most important structural differences. The analysis carried out, with the work-flow that will be explained in the next paragraphs, allowed acquisition of useful information that will be collected in a scientific work.
Due to the high interest of the scientific community on this target, we have already produced a manuscript entitled “Computational Studies of SARS-Covid2 3CLpro: Insights from MD Simulations”, that will be submitted to the International Journal of Molecular Sciences - Special Issue “Exscalate4CoV: Innovative High Performing Computing (HPC) Strategies to Tackle Pandemic Crisis” in a few days. In this paper we discuss the main differences coming from the analysis of the whole protein structural behavior and those seen in the binding site. The web address of the repository will be communicated shortly. It will be possible to download the trajectories of the simulations discussed in the work.
Papain-like proteinase (PL-PRO): Responsible for the cleavages located at the N-terminus of the replicase polyprotein. In addition, PL-PRO possesses a deubiquitinating/deISGylating activity and processes both 'Lys-48'- and 'Lys-63'-linked polyubiquitin chains from cellular substrates. Participates, together with nsp4, in the assembly of virally induced cytoplasmic double-membrane vesicles necessary for viral replication. In the video, the PL-PRO is shown in green highlight.
Nsp12-7-8: A key component, RNA-dependent RNA polymerase [RdRp, also known as nsp12], catalyzes the synthesis of viral RNA, and therefore plays a central role in the replication and transcription cycle of the COVID-19 virus, possibly with the help of nsp7 and nsp8 as cofactors. In the video, the nsp12/nsp7/nsp8 hetero-oligomeric complex is shown in highlight. Nsp12, nsp7 and nsp8 are shown in blue, red and green respectively.
<Nsp13: Scientists suspect that nsp13 unwinds so that other proteins can read its sequence and make new copies. This protein, called Helicase, is a multi-functional protein with a zinc-binding domain in the N-terminus displaying RNA and DNA duplex-unwinding activities with 5' to 3' polarity. Activity of helicase is dependent on magnesium. Here, the protein is reported in blue highlight.
<Nsp15: This enzyme is a specific endoribonuclease with a C-terminal catalytic domain, belonging to the EndoU family. EndoU enzymes are present in all animal kingdoms, where they perform various biological functions associated with RNA processing. Researchers suspect that this protein cuts the residual virus RNA as a way of hiding from the antiviral defenses of the infected cell. The protein, in its hexameric form, is shown in the video shows in highlight style, and each monomer composing the hexamer has a different color.
Spike receptor-binding domain (RBD)/ACE2: Dynamic structure of the receptor-binding domain (RBD) of the spike protein of SARS-CoV-2 bound to the cell receptor ACE2. Coronaviruses use the spike glycoprotein on the envelope to bind to their cellular receptors. Such binding triggers a cascade of events that leads to the fusion between cell and viral membranes for cell entry. The video shows in highlight, the SARS-CoV-2 RBD core in slate and ACE2 in red.
CADD
Summary
A systematic mapping of the druggable cavities within the SARS CoV-2 therapeutically relevant proteins.
This study provided a novel strategy for pocket-mapping based on the combination of pocket (as performed by the well-known FPocket tool) and docking searches (as performed by PLANTS or AutoDock/Vina engines). Such a mapping enables the identification of the most relevant binding sites for which virtual screening simulations or de novo rational design should allow the identification of promising hits.
A few more details
Such an approach is implemented by the Pockets2.0 plugin for the VEGA suite of programs. The VEGA suite comprises a graphical interface with a new version of the plug-in for FPocket (named Pockets2.0), a well-known software used to detect protein cavities, based on an optimized algorithm for Voronoi tessellation. For a better exploration of the protein cavities, this combines the already implemented cavity mapping, as performed by Fpocket, with docking calculations with probe molecule(s) using AutoDock/Vina or PLANTS docking programs. To optimize the ranking of the explored cavities, Pockets2.0 can utilize both Fpocket and docking scores by calculating customizable consensus scores. The combination of the FPocket and docking scores by calculating customizable consensus scores leads to a significant increase of the correctly identified binding sites compared to the FPocket and docking scores alone, and this enhancement appears to be truly relevant when analyzing complex proteins with rather narrow binding pockets, and in particular, for characterizing allosteric binding sites.
The scientific paper entitled “A systematic mapping of the druggable cavities within the SARS CoV-2 therapeutically relevant proteins by combining pocket and docking searches as implemented in Pockets2.0”has been submitted to International Journal of Molecular Sciences - Special Issue “Exscalate4CoV: Innovative High Performing Computing (HPC) Strategies to Tackle Pandemic Crisis”. The web address of the repository will be communicated shortly. It will be possible to download all the structural data discussed in the work.
The images below show the process of the viral protein mapping (Figure 1) and the identification of the binding pocket (Figure 2). In particular, figures 1 and 2 represent the homologic 3D structures of the viral protein nsp13 and the heteromer nsp14-nsp10 respectively, generated in the deliverable D1.1. This process underlines the importance of collaboration among the activities carried out by each partner.
CADD
Summary
Machine Learning and Virtual Screening (VS) protocol optimization and Drug Repositioning (D1.3, D1.4, D1.5).
Several docking simulations were performed to define and optimize the machine learning and virtual screening protocols to use on SARS CoV-2 proteins. The performances of the virtual screening strategies were assessed by evaluating their capacity to correctly rank molecules, which are endowed with antiviral activity, and in particular, with a known effect against SARS CoV proteins, considering the lack of known actives on SARS CoV-2 proteins.
The tuned and validated virtual screening protocols were used to screen a repurposing library, containing the set of safe in man drugs, commercialized or under active development in clinical phases, and a set of known bioactives in particular preclinical compounds identified as “CoV Inhibitors” (> 12000 drugs). The most promising drugs, identified from docking studies as potentially active against SARS-Cov-2 proteins, were selected to be tested in biochemical and phenotypic assays performed respectively in WP2 and WP3.
A few more details
The SARS CoV-2 3CL-Protease was analyzed and used to fine tune the virtual screening strategies. SARS CoV 3CL-Protease known inhibitors were collected from literature to define training sets for our studies, keeping in mind that the pocket of SARS-Cov and SARS-Cov-2 proteases are very similar. Different docking algorithms were combined to increase the quality of the virtual screening campaign.
Five consortium partners (Dompé, University of Milan, and the three supercomputing centers, CINECA, Barcelona Supercomputing Center (BSC) and Jeulich Forschungszentrum (FZJ)), made their hardware and software facilities available to run docking simulations, including LiGen, PLANTS, PELE, Glide and FRED. The virtual screening protocols were validated in terms of Enrichment Factor, setting the conditions that allow most of the training set compounds to be ranked in the top-scored positions. Subsequently, the molecules with the best docking scores, found in the top 1% of the screened library, were selected with each software, and a consensus approach of the different docking scores was used to rank the most promising safe in man drugs acting on 3CL-PRO viral protein (Figure 1). To further improve the quality of the results, more accurate methods were applied, such as induced-fit simulations on SARS-CoV known inhibitors with PELE software, and a regression analysis model to correlate predicted and in vitro activities was built. We obtained a clear correlation of higher IC50 with larger residence time in the active site, which was analyzed in terms of (1) the solvent-accessible surface area of the ligands, (2) the sub-pocket population analysis. Both analyses showed high potential for a more accurate prediction, opening the possibilities, for example, to a second refinement effort after an initial docking campaign.
We are now using the tuned and validated virtual screening protocols to screen the drug repurposing library against the best selected 3D structures of the other SARS-Cov-2 proteins, either already experimentally obtained (PDB) or derived by homology modelling techniques, and processed by molecular dynamics simulation (Figure 2).
We currently have validation compounds coming from the literature, and results coming from our docking approaches that identified different sets of molecules. We obtained a critical assessment of different approaches and results that we can get from combining the best of the techniques we have available and that we are putting in place, for the identification of the most promising safe in man drugs that are ready for immediate treatment of the infected population.
PROTEIN PRODUCTION AND TARGET BASED ASSAYS
Summary
Identification of inhibitors of SARS-CoV-2 3CL-Pro enzymatic activity using a repurposing screen (“target confirmation of main protease”)
SARS-CoV-2 main protease (3CL-Pro), also termed M-Pro, is an attractive drug target as it plays a central role in viral replication by processing the viral polyproteins pp1a and pp1ab at multiple distinct cleavage sites. Within our repurposing program we confirmed previously reported inhibitors of 3CL-Pro and have identified 62 additional compounds with IC50 values below 1 μM. A subset of eight inhibitors showed anticytopathic effect in a Vero-E6 cell line. , and the X-ray crystal structure of the complex of one of the inhibitors and SARS-Cov-2 3CL-Pro was solved, showing covalent binding to the catalytic Cys145.
PROTEIN PRODUCTION AND TARGET BASED ASSAYS
Summary
X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease (“How do you find 37 new repurposed drugs that have direct 3D binding to SARSCoV2's main protease to knock out its ability to replicate”)
In a search for a drug against COVID-19, we have performed a high-throughput X-ray crystallographic screen of two repurposing drug libraries against the SARS-CoV-2 main protease (Mpro), which is essential for viral replication. In contrast to commonly applied X-ray fragment screening experiments with molecules of low complexity, our screen tested already approved drugs and drugs in clinical trials. From the three-dimensional protein structures, we identified 37 compounds that bind to Mpro. In subsequent cell-based viral reduction assays, one peptidomimetic and six non-peptidic compounds showed antiviral activity at non-toxic concentrations. We identified two allosteric binding sites representing attractive targets for drug development against SARS-CoV-2.103 authors mainly from science organizations in Hamburg contributed to this work; 5 of our ITMP scientists contributed scientific input and data interpretation and managed to supply our compounds to this X-ray crystallography screening.
PHENOTYPIC SCREEN
No results yet
STRUCTURAL BIOLOGY
Summary
The structural biology team of Elettra Sincrotrone Trieste has focused on SARS-CoV-2 Mpro viral protein by setting up a reproducible expression, purification and crystallization protocol and using thermal stability profiling to define protein quality control and batch to batch reproducibility. Thousands of protein crystals of the APO and HOLO forms have been obtained, and tested at the XRD2 beam line of Elettra. Best crystals diffracted at a resolution of 1.3 Å. Hundreds of data set have been collected, processed and refined and resulting 3D models confirmed the Mpro dimeric structure of with its typical fold.
Figure 1 shows examples of Mpro crystals and diffraction patterns, Figures 2 and 3 show the 3D graphic representations of the Mpro dimer.
A few more details
The structural biology team of Elettra Sincrotrone Trieste involved in the WP4, started to work in April 2020, anticipating the planned activities by about 5 months with respect to the original schedules. The initial effort has been dedicated to protein expression, purification and sample characterization with crystallization screening set-up.
The SARS-CoV-2 Mpro was successfully expressed in E.coli and purified at homogeneity (> 98% purity) starting from the expression vector kindly donated by L. Hilgenfeld (Lubeck University – DE). Initial crystallization screenings have been set up based on published data and using commercially available crystallization kits. The most promising conditions have been optimized and “flower-like” shape crystals were reproducibly obtained. These crystals were cryo-preserved and tested at the XRD2 beamline of the Elettra synchrotron, showing a diffraction pattern with a resolution in the range of 1.6-2.0 Å, with the best resolution achieved at 1.52Å. Datasets were processed and a quick MR solution was done using pdb 6W63 as a starting model showing either P21 or C2 space group, having a monomer or a dimer in the asymmetric unit, respectively, but with almost identical conformation. Further optimization of the crystallization trials has established also a seeding based protocol that grew crystals of space group P212121. For each space group we obtained the crystal structure of Mpro in APO form and the coordinates have been deposited in PDB with code 7ALI (https://www.rcsb.org/structure/7ALI, sp.gr. P21), 7ALH (www.rcsb.org/structure/7ALH, sp.gr. C2) and 7BBE (www.rcsb.org/structure/7BB2, sp.gr. P2(1)2(1)2(1)).
Xray diffraction studies on crystals of Mpro bound to specific inhibitors have been performed. A number of small molecules selected from a biochemical screening of 8700 repurposing drugs have been tested by co-crystallization and soaking techniques. So far 8 structures have been deposited in PDB describing the binding modes of myricetin, Ebselen, SU3327, MG-132 in absence and in presence of DTT. Binding mode of myricetin, a flavonoid, has been clearly identified in the crystal structure (Fig. 4) showing a covalent linkage with the catalytic cys145. The results have been published in two papers confirming the findings of the biochemical screening [Kuzikov et al. 2021; doi.org/10.1021/acsptsci.0c00216] and validating the outputs of virtual screening and molecular dinamics [Gossen et al. 2021; https://doi.org/10.1021/acsptsci.0c00215]. The coordinates of the crystal model are deposited in PDB with code 7B3E (www.rcsb.org/structure/7B3E). All structures in PDB have been released immediately and are openly available.
Deposited PDB:7ALH, 7AL, 7B3E, 7BB2, 7BE7, 7BFB, 7BGP, 7NBY, 7NF5, 7NG3, 7NG6
GENOMICS
Summary
Main activities performed in WP5 include the use of techniques for molecular evolution, phylogenetic and multiple correspondence analysis (MCA) for the identification of genomic and protein sites associated with SARS-CoV-2 adaptation and host-pathogen interaction.
As a first step, the analysis focused on the spike protein from SARS-CoV-2 and other representative β-Coronaviruses. Our results shows that the unsupervised analysis of conservation patterns across the β-CoV spike protein family can help tracing the amino acid space encoding the specificity of β-CoVs to their cognate host cell receptors. More precisely, our results indicate that host cell receptor usage is encoded in the amino acid sequences of different CoV spike proteins in the form of a set of specificity determining positions (SDPs)1.
A few more details
Here, we aim at analyzing the effect of evolutionary constraints in shaping the organization of the spike protein family across the β-CoV lineage. To this aim, we use a multivariate analysis protocol for unsupervised detection of both the protein family segregation and the residue positions that better explain the sources of variation of the family, i.e. Specificity Determining Positions (SDPs).
The analysis was performed both at the level of the full β-CoV genus and individual subgroups. While in the first case the results are consistent with the known phylogenetic classification of Betacoronavirus into five subgenera (Fig. 1C), the analysis performed on the individual β-CoV groups allowed a fine-grained separation into clusters that clearly reflect the functional diversification of the spike protein family, that is, the specificity to different host-cell receptors. Indeed, both the clustering and domain enrichment results of the identified SDPs consistently reproduce the known cell receptor specificities observed across the different β-CoV lineages. At the level of the Sarbecovirus group, for example, both SARS-CoV-2 and RaTG13 are clustered together with SARS-CoV and other SARS-like sequences from bats (Fig. 1A,D), reflecting the ability of these members of the Sarbecovirus group to bind the ACE2 cell receptor. Notably, the proximity of SARS-CoV-2 and RaTG13 and other SARS-CoV sequences based on the SDPs is different to what observed based on phylogenetic analysis, where they form distinct clades.
Collectively, our results point to a key role of the SDPs in mediating host cell receptor specificity across β-CoVs and provide, at the same time, a framework for monitoring the evolution of the SARS-CoV-2 specificity to hACE2, as well as the emergence of novel potential cross-species transmission events. As such, it is worth mentioning that from the analysis of amino acid variations across the circulating SARS-CoV-2 virus, SDPs found in the RBD tend to mutate with a very low frequency, similar to what is seen at ACE2 contacting sites. This is of relevance, as our results suggest that mutations in SDPs can significantly impact the receptor-binding ability of the spike. In line with this reasoning, it is important to notice that other members of the Sarbecovirus group might have the potential to acquire ACE2 binding ability, as they share substantial similarity in terms of SPDs. This is especially the case of members of the Sarbecovirus clade 3, which despite being phylogenetically distant to SARS-CoV-2, display identical residues in 10 out of 14 SDP positions within the RBD, making them potential candidates for new human infections.
In summary, our results show that the identification of evolutionary patterns based on the analysis of sequence information alone can provide meaningful insights on the molecular basis of host-pathogen interactions and adaptation. Both the methodology and results obtained in this work lead the basis for a follow-up study where, in collaboration with other members of the E4C consortium, we aim at identifying potential routes of mutations that could lead to new β-CoVs adaptation to human hosts, and ultimately contribute to better understanding and monitoring of events that are critical to public health concerns worldwide.
1Pontes C, Ruiz-Serra V, Lepore R, Valencia A. Unraveling the molecular basis of host cell receptor usage in SARS-CoV-2 and other human pathogenic β-CoVs. Comput Struct Biotechnol J. 2021;19:759-766. doi: 10.1016/j.csbj.2021.01.006
In parallel, continuous monitoring of the virus evolution is performed based on the analysis of the SARS-CoV-2 genomic sequences from public repositories. Variation analysis is performed based on the established SARS-CoV-2 NC_045512.17 as a reference genome, where amino acid variations are annotated for all major coding regions as well as alternative reading frames and mutation frequencies computed globally and by different countries at different time points. Data are continuously updated as new sequences become available and shared with the partners and the community via the ViralSeq website
MECHANISM OF ACTION
No results yet
AUTOMATED HOMOLOGY MODELING WORKFLOW FOR DRUG TARGET
No results yet
PRODUCTION AND TUNING ON HPC INFRASTRUCTURE
Summary
In the WP8 “Production and tuning on HPC infrastructure”, the POLIMI team is responsible for the continuous tuning and code adaptation of the EXSCALATE platform, and contributes to its porting on the new Marconi100 partition at CINECA.
The first month of the E4C project coincided with the initial production period of the new CINECA machine. POLIMI ported a first version of the docking library to exploit the computing capability of the heterogeneous computing node composed of 2xIBM-Power9 Sockets and 4xNVidiaV100.
During the experimental campaign, we reached a throughput of more than 250K optimal ligand poses per second on a single node of the Marconi100 machine. Porting and tuning on multiple nodes is currently ongoing.
A few more details
To promote agile and portable software development needed to guarantee continuous releases of updated functionalities and improved elaborated throughput of the EXSCALATE software platform, we encapsulated all the geometrical docking, issued in a stand-alone library called LiGen GeoDock, with a simple, stable, and well-defined non-virtual interface.
Figure 1 shows an overview of the LiGen GeoDock library, which aims at docking a ligand in a target pocket using geometrical information only. To better encapsulate Geodock in the EXSCALATE platform, we designed a single public interface hiding implementation details. In this way, we can improve GeoDock without hindering the development of the other components. The E4C project targets a heterogeneous node composed of GPUs and CPUs. The previous C++ code version was unable to harness the full computational power of the new CINECA Marconi100 node based on CPUS only. To overcome this limitation, we implemented the docking algorithm using the OpenACC pragma-based language and the PGI OpenACC compiler to generate device-specific code while maximizing the code portability. However, given the transition to the Marconi100 supercomputing cluster at CINECA, we then implemented the docking algorithm using the CUDA C/C++ language to obtain the maximum performance from of the NVIDIA Tesla V100 GPUs.
We ran an initial experimental campaign to assess the ligand library performance with a wide range in the number of atoms and rotatable bonds. The code peak performance using only the 2xIBM-Power9 and 128 software threads reaches a throughput of 30K optimal ligand poses per second, while the exploitation of the full node including the 4xNvidiaV100 GPUs reaches a throughput of more than 250K optimal ligand poses per second.
These performance results obtained so far on the new CINECA Marconi100 partition are very promising, and they are a good starting point for further tuning of the LiGen GeoDock library. As next steps, we envision pushing further on a fine-grain refinement and adopting dynamic autotuning approaches, combined with the entire machine-level scaling.
REGULATORY CONTRACTS
Raloxifene
Raloxifene has emerged from the Exscalate platform as a molecule with potential effectiveness against SARS-CoV-2, based on antiviral targeting. This molecule is used as active ingredient in medicines already authorized on the European Community for the treatment of post-menopausal osteoporosis. A preliminary development and registration strategy was shared with the European Medicines Agency (EMA) in October 2020 through a procedure called Rapid Scientific Adivice.
The product is currently being used in a placebo-controlled Phase 2/3 clinical study, which is ongoing in Europe.
DISSEMINATION & EXPLOITATION OF THE FOREGROUND
Summary
Setup and Maintenance of Website & Social Media (D10.1).
Chelonia leads the coordination of dissemination and communication a
ctivities. The project webpage www.exscalate4cov.eu was produced within the first month of the project and is now regularly updated and maintained. Communication activities ensure the diffusion of the project outcome outside the Consortium so that the methodologies developed and the results obtained can be widely disseminated in the scientific community.
Communication objectives are achieved by using multiple different strategies including the use of social networks such as Linkedin, Facebook and Twitter to spread the project knowledge to a larger audience.
Dedicated exscalate4cov pages have been created and all communication are mainly tagged #exscalate4cov. Given the wide social impact of this research, actions towards the general press are given through press releases, newsletters and through local press.
DISSEMINATION & EXPLOITATION OF THE FOREGROUND
Summary
Initial Dissemination Activities and Plans (D10.2).
This task defines the strategy for exploitation of the project results. The activity, led by CHELONIA, in tight collaboration with DOMPE, analyzes the strategies and implements the actions to maximize the utilization of the project results, beyond the project partners and time-frame. A dissemination plan was issued to all partners 3 months earlier than the expected deadline in order to facilitate dissemination and to align partners in communicating projects results to the target audience. A first webinar was organized on April 28th, 2020, thanks to the support of CECAM (Centre Européen de Calcul Atomique et Moléculaire – www.cecam.org ), which is an organization devoted to the promotion of fundamental research on advanced computational methods and to their application to important problems in frontier areas of science and technology. To access the recorded webinar please visit YOUTUBE


















