A Comprehensive Mapping of the Druggable Cavities within the SARS-CoV-2 Therapeutically Relevant Proteins by Combining Pocket and Docking Searches as Implemented in Pockets 2.0
Gervasoni, S.; Vistoli, G.; Talarico, C.; Manelfi, C.; Beccari, A.R.; Studer, G.; Tauriello, G.; Waterhouse, A.M.; Schwede, T.; Pedretti, A. A Comprehensive Mapping of the Druggable Cavities within the SARS-CoV-2 Therapeutically Relevant Proteins by Combining Pocket and Docking Searches as Implemented in Pockets 2.0. Int. J. Mol. Sci. 2020, 21, 5152.
ABSTRACT
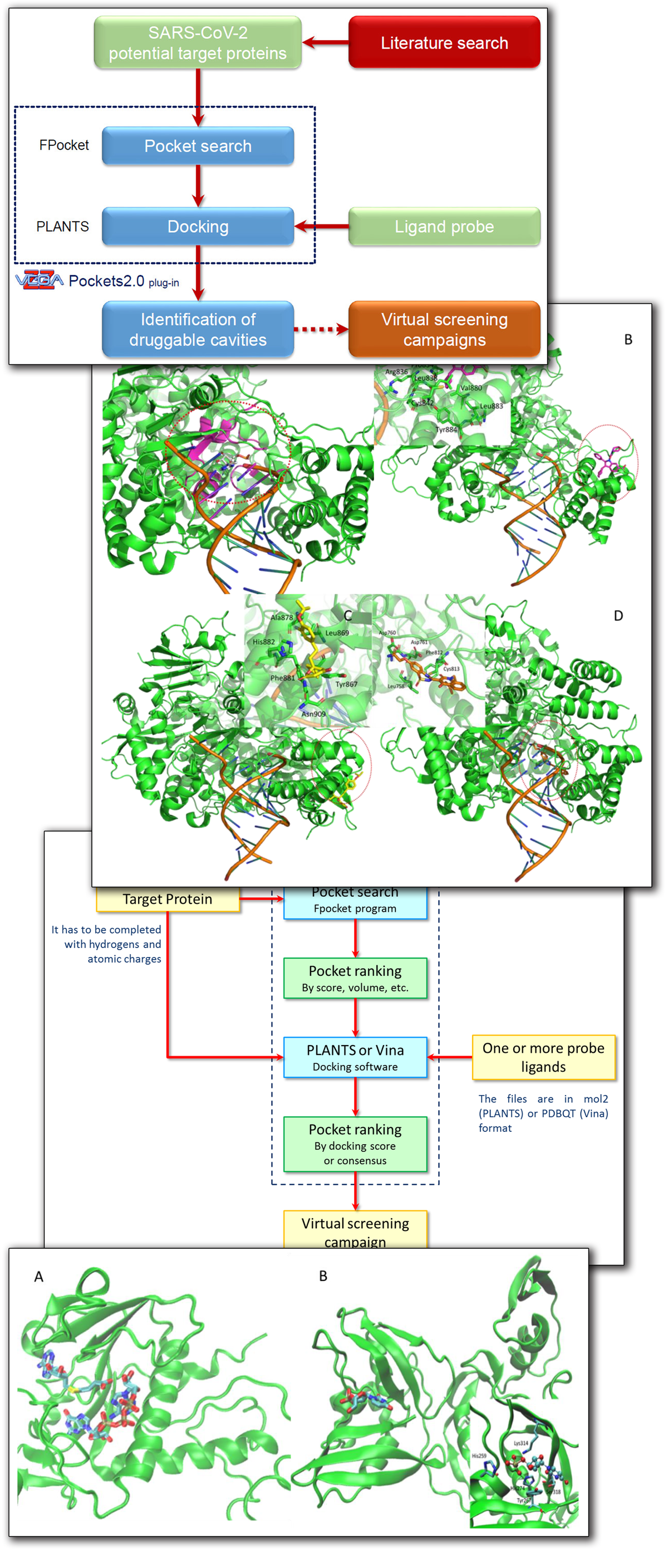
(1) Background: Virtual screening studies on the therapeutically relevant proteins of the severe acute respiratory syndrome Coronavirus 2 (SARS-CoV-2) require a detailed characterization of their druggable binding sites, and, more generally, a convenient pocket mapping represents a key step for structure-based in silico studies; (2) Methods: Along with a careful literature search on SARS-CoV-2 protein targets, the study presents a novel strategy for pocket mapping based on the combination of pocket (as performed by the well-known FPocket tool) and docking searches (as performed by PLANTS or AutoDock/Vina engines); such an approach is implemented by the Pockets 2.0 plug-in for the VEGA ZZ suite of programs; (3) Results: The literature analysis allowed the identification of 16 promising binding cavities within the SARS-CoV-2 proteins and the here proposed approach was able to recognize them showing performances clearly better than those reached by the sole pocket detection; and (4) Conclusions: Even though the presented strategy should require more extended validations, this proved successful in precisely characterizing a set of SARS-CoV-2 druggable binding pockets including both orthosteric and allosteric sites, which are clearly amenable for virtual screening campaigns and drug repurposing studies. All results generated by the study and the Pockets 2.0 plug-in are available for download.